All published articles of this journal are available on ScienceDirect.

Role of Genes and Treatments for Parkinson’s Disease
Abstract
Parkinson’s Disease (PD) is a complex neurodegenerative disorder that mainly results due to the loss of dopaminergic neurons in the substantia nigra of the midbrain. It is well known that dopamine is synthesized in substantia nigra and is transported to the striatum via nigrostriatal tract. Besides the sporadic forms of PD, there are also familial cases of PD and number of genes (both autosomal dominant as well as recessive) are responsible for PD. There is no permanent cure for PD and to date, L-dopa therapy is considered to be the best option besides having dopamine agonists. In the present review, we have described the genes responsible for PD, the role of dopamine, and treatment strategies adopted for controlling the progression of PD in humans.
1. INTRODUCTION
Neurodegeneration refers to the condition which primarily affects neurons [1]. Hippocrates (460-375 BC), in his important treatise entitled ‘On The Sacred Disease’ describes epilepsy and relates it with the brain. In the ancient period, neurological disorders were classified under humor- Vata rogas (diseases) in Ayurveda. Vata refers to the energy present in the body involved in controlling both voluntary and involuntary functions. Any change in vata leads to the pathogenesis of the neurodegenerative disease. In Ayurveda, about eighty types of neurological disorders have been described, such as apasmara /apasmati (Epilepsy), kampavata (Parkinson’s disease). The clinical symptoms and treatments of neurodegenerative disease have been described by Hippocrates [2-5].
The clinical symptoms of neurodegenerative diseases are often characterized by alteration in cognitive function, movement, memory and learning, co-ordination and autonomic control [6]. Worldwide neurological disorders have more than 6% of the total burden of diseases [7]. The most common neurological diseases are Alzheimer's Disease (AD), Parkinson’s Disease (PD), Huntington's Disease (HD), Amyotrophic Lateral Sclerosis (ALS), fronto-temporal dementia and the spinocerebellar ataxia (Table 1).
Neurodegenerative diseases/disorders have different pathophysiology, along with cognitive impairments. The loss of brain cells in neurodegenerative diseases is not a rapid process but rather a very slow process that becomes worse during the progression of the disease. Sporadic, as well as familial forms of neurodegenerative diseases, are common in the human population. These forms are difficult to distinguish phenotypically as well as histologically. A number of causative genes have been discovered for the neurodegenerative disease [8]. James Parkinson was born on April 11, 1755, in the semi-urban hamlet of Hoxton, Northeast of the city of London, England. He describes PD for the first time. PD is an age-progressive disease, (second after AD), of the nervous system. James Parkinson was the first who provided a description of “Paralysis agitans” or “Shaking palsy”, in his popular monograph named “An Essay on the Shaking Palsy”, in 1817 [9, 10].
2. BASIC STRUCTURE AND FORMATION OF DOPAMINE IN DOPAMINERGIC NEURONS
PD is a complex neurological disorder of CNS that affects mainly the motor system of the body. The non-motor symptoms also become worsen as the disease progresses [11, 12]. Early symptoms of the PD are shaking, rigidity, slowness of movement, and difficulty in walking [13, 14]. The behavioural problems may also occur with the disability of thinking power. It is reported that about 3% of the affected
| Neurodegenerative Diseases | |
| Diseases | Characteristics |
|
Dementia Dr. Philippe Pinel, 1797 |
The two lobes become damaged and then shrink. The genetic mutations are thought to have a negative effect on a protein called the tau protein. All brain cells contain tau proteins that help to keep them stable but abnormal tau proteins may lead to dementia. |
|
Parkinson's disease (PD) Dr. James Parkinson, 1817 |
Normally brain neurons produce chemical messenger known as dopamine. When the neurons die or become impaired, they produce less dopamine and Lewy bodies formed unusual clumps of the protein α-synuclein in substantia nigra which causes the PD. |
|
Spino-cerebellar Ataxia (SCA) Dr. Nikolaus Friedreich, 1863 |
Mutation in several forms of SCA1 protein in brainstem cerebellum and spinal cord lead to Spino-cerebellar Ataxia. |
|
Huntington's disease (HD) Dr. George Huntington, 1872 |
Mutation in HTT gene instruct to make a protein i.e. huntingtin. DNA segment “CAG” segment is repeated normal 36 to more than 120 times which cause HD. |
|
Amyotrophic lateral sclerosis (ALS) Dr. Jean-Martin Charcot, 1874 |
Mutations in gene encoding superoxide dismutase 1 (SOD1) in nerve cells that cause ALS. |
|
Alzheimer's disease (AD) Dr. Alois Alzheimer, 1906 |
Abnormal clumps i.e. amyloid plaques and tangled bundles of fibers i.e. neurofibrillary tangles or tau. |
people are above 65 years of age [15]. In PD, nerve cells of substantia-nigra of the mid-brain are degenerated. The basal ganglia help in initiating and smoothening of voluntary muscle movements, suppresses involuntary movements and problem in the coordination in changes of posture [16, 17]. Each part of the brain consists of Globus pallidus i.e. internal and external. The other part of the brain which is involved in the neurotransmitter function is the striatum, which includes caudate nucleus, putamen and substantia nigra. There are basically two pathways by which three main neurotransmitters work i.e. Glutamate, GABA and Dopamine [18, 19]. The basal ganglia release neurotransmitters (chemical messenger) for the activation of nerve impulse pathways. The main neurotransmitter in the basal ganglia is dopamine [20]. If the nerve cells in the basal ganglia are degenerated then there will be less production of dopamine, leading to the loss of control on muscle movements (Fig. 1). It leads to tremors, bradykinesia, hypokinesia, posture problems and the loss of overall coordination of functions. The substantia nigra, which is degenerated in PD plays an important role in the normal functioning of motor and non-motor activity of the body. The substantia nigra sends chemical message through axons that pass throughout the striatum and help in the movement of a particular part of the body. This is known as the nigrostriatal pathway or dopamine secretion pathway [21, 22].
The signals from one neuron to another are transferred through the active neuron refers as dopaminergic neurons. Within the striatum, there are neurons having specific receptors to recognize and respond to dopamine only. These receptors cannot be activated by any other neurotransmitters. When the cells in substantia nigra are dead, the level of dopamine decreases rapidly as a result, striatal cells are not activated and the PD symptoms increases day by day [23, 24]. The presence of dopaminergic neurons in the brainstem was first reported by Dahlstrom and Fuxe in 1964. In the pathophysiology of PD, the importance of dopamine depletion was first suggested by Carlsson in the late 1950s [25, 26]. In 1960, Ehringer and Hornykiewicz published their report after examining six cases of PD that the dopamine content in the neostriatum was found to be severely reduced. Dopamine (an amine), is synthesized by a precursor molecule L-Dopa. It is synthesized in the brain as well as in kidneys [27, 28].
It is well established that dopamine itself is a neurotransmitter that is synthesized in substantia nigra and transported to the striatum via the nigrostriatal tract. Several other functions have also been attributed to dopamine i.e. it can act as a vasodilator, it enhances sodium excretion and urine output; it reduces insulin production, gastrointestinal motility and protects intestinal mucosa. It also reduces the activity of lymphocytes [25, 29]. Dopamine is not able to cross the blood-brain barrier, so the interest was focused on the dopamine precursor levodopa. Levodopa takes entry into the brain with a large neutral amino acid transport pathway and can be decarboxylated to form dopamine. Henry Dale, a Physician, was first to examine the biological activity of dopamine in 1910. He suggested the shorter name dopamine in 1952. The Physician George Barger and James Hill synthesized 3,4-dihydroxyphenylethylamine at The Well-Come Laboratories in London [30]. At the same time, Kathleen Montagu and Arvid Carlsson, together with Lindqvist, Magnusson, and Waldeck in 1957, reported the presence of dopamine in the brain, and in 1959, the Swedish group claimed the presence of dopamine in high concentrations in the striatum [31-33]. Marthe Vogt suggests that there is a continuous discharge of dopamine but was not sure about dopamine to be a conventional neurotransmitter in the neurons of nigrostriatal regions [34]. Dahlstrom (1973) [35] was also not sure about the presence of dopamine in the Central Nervous System (CNS). Dopamine receptors play an essential role in daily life functions. There are various dopamine receptors in the CNS, specifically in the hippocampal dentate gyrus and sub-ventricular zone. The five types of dopamine receptors, namely, D1, D2, D3, D4, and D5 has been described to date. Each receptor has a different function. According to Mishra et al. (2018) [36] the functions of various dopamine receptors are as follows:
- D1: memory, attention, impulse control, regulation of renal function and locomotion.
- D2: locomotion, attention, sleep, memory and learning.
- D3: cognition, impulse control, attention and sleep.
- D4: cognition, impulse control, attention and sleep.
- D5: decision making, cognition, attention and renin secretion.
Further, the two types of sub-families of dopamine receptors are D1-like family and D2 like family. The D1 and D5 receptors belong to D1-like family of dopamine receptors, and D2, D3 and D4 receptors belong to the D2-like family [37]. D1 and D5 receptors coupled to G stimulatory sites and activate adenylyl cyclase. The activation of adenylyl cyclase leads to the production of the second messenger cAMP, which further leads to the production of protein kinase A (PKA) and affects transcription in the nucleus [38, 39]. The D1 receptor is most abundantly distributed in the CNS, followed by D2, D3, D5 and D4 (D1>D2>D3>D5>D4) [40]. D1 receptors regulate the development of neurons are play an important role in the regulation of most of the cognitive activities as well as locomotion [41]. D1 and D5 receptors have high density in the striatum, nucleus accumbens, olfactory bulb, and substantia nigra. D1 and D5 receptors, along with stimulating adenyl cyclase, also activates phospholipase C, which leads to the induction of intracellular calcium release and activation of protein kinase C. Protein kinase C is a calcium-dependent protein kinase. Calcium is also involved in modulating the neurotransmitter released by exocytosis. D1 and D5 inhibits Na/K ATPase i.e. cyclic AMP-dependent Protein Kinase (PKA) calcium- and phospholipid-dependent Protein Kinase (PKC) pathways in the kidneys [42]. D2, D3, and D4 receptors are expressed mainly
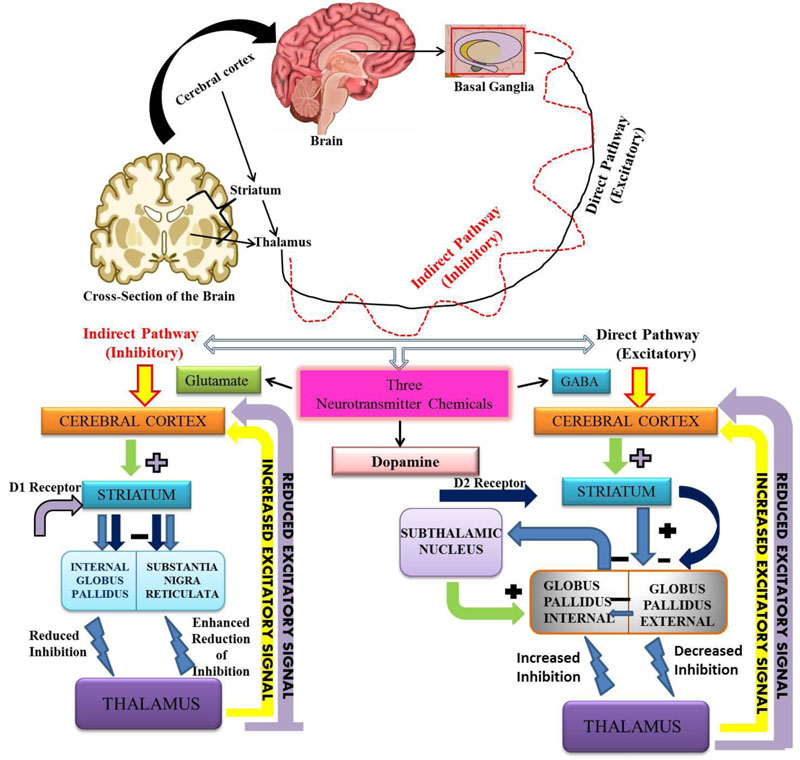
in the striatum, external globus pallidus, core of nucleus accumbens, hippocampus, amygdala, and cerebral cortex. These receptor affects the postsynaptic receptor-mediated extrapyramidal activity. D2-D4 receptors are important for signaling, the survival of human dopaminergic neurons and their development [43, 44]. It has been reported in mice that the dysfunction of the dopaminergic pathways leads to neurological and psychiatric disorders [45, 46]. Fig. (2) shows a comparison of the normal brain with a brain affected by various factors that are responsible for inducing PD. The dopamine signaling is important for maintaining the normal physiological processes. The disruption of pathways may lead
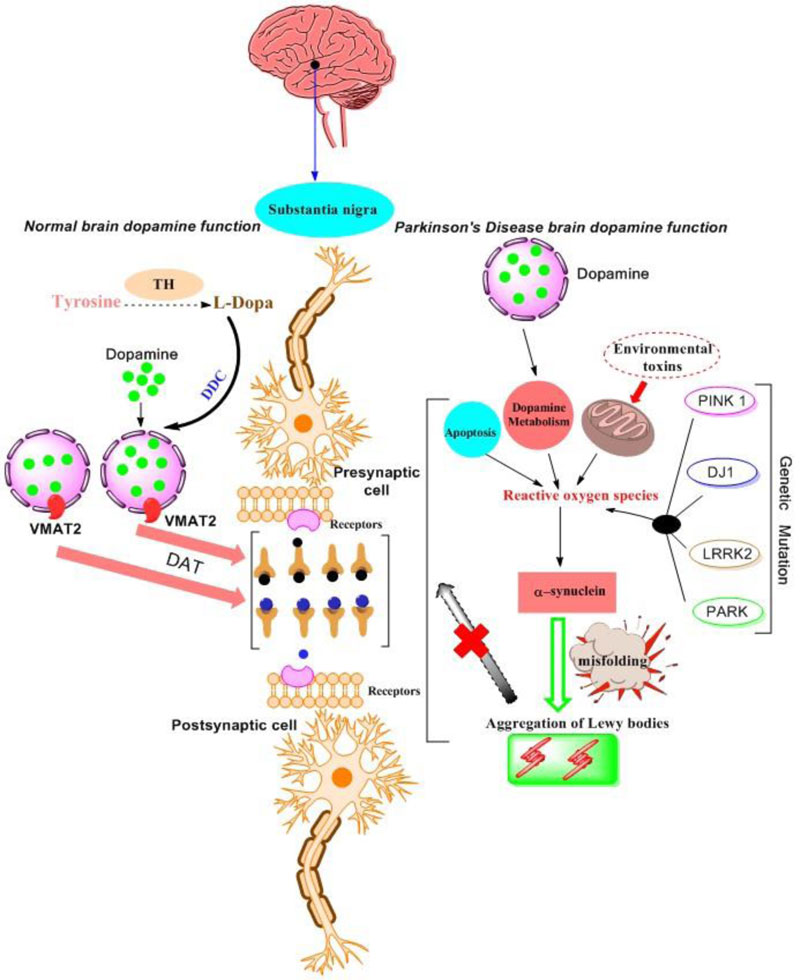
to the dysfunctions attributed to neurodegenerative disorders. Tyrosine in the presence of Tyrosine Hydroxylase (TH) is converted into L-Dopa. Then, L-Dopa is converted into the Dopamine in the presence of DOPA Decarboxylase (DDC). Dopamine is transported from the cytosol by a Vesicular Monoamine Transporter (VMAT2) into the synaptic vesicles where it is stored until its release into the synaptic cleft. Dopamine receptors are present in both post as well as pre-synaptic neurons (including Dopamine Transporter, DAT). The required dopamine reaches the postsynaptic synapse through the dendrites, which leads to the normal functioning of both motor and non-motor neurons [47].
3. GENETICS OF PARKINSON’S DISEASE (PD)
Several common cellular mechanisms of protein aggregation are involved in the progression of neurodegenerative diseases. The β-amyloid is the main part of plaques formation in AD, α-synuclein for Lewy bodies (LBs) formation in the substantia nigra in PD and dementia with LBs (DLB), and expanded repetition of polyglutamine is found as intra-nuclear and cytoplasmic inclusions in HD [48, 49]. The LBs were first described by F.H. Lewy in 1912, in the nucleus of Basalis of Meynert and the dorsal vagal nucleus in patients with PD. It was named in the honour of Friederich H. Lewy by Tretiakoff, who confirmed the presence of LBs in the substantia nigra [50-52]. LBs are rounded, eosinophilic and intra-cytoplasmic neuronal inclusion or aggregation of α-synuclein. According to some studies, the LBs are found in the selected areas of the brain i.e. substantia nigra, locus ceruleus, dorsal vagal nucleus, the nucleus of Meynert, hypothalamus cortex and sympathetic ganglia [53-55]. For the last 20 years several important discoveries have been made by researchers on the genetics of PD. The SNCA and its mutations are associated with autosomal dominant parkinsonism [56, 57]. Mutations in this gene include missense, which results in amino acid substitutions. SNCA-related PD is not so common, but identification of SNCA mutations has led to the identification of α-synuclein as the major component of Lewy bodies [58-61]. α-synuclein has 140 amino acids, that belongs to the family of related synucleins (β-synuclein of 134 amino acids) [62, 63]. α-synuclein contains an acidic stretch towards the C-terminus and there are no cysteines and tryptophans. Structurally, human α-synuclein consists of three regions i.e. an N-terminal amphipathic region, a hydrophobic central region, and an acidic C-terminal region [61, 64-66]. Six genes have been found to be associated with autosomal dominant forms of PD i.e. SNCA, LRRK2, VPS35, EIF4G1, DNAJC13, and CHCHD2 and three with autosomal recessive forms of PD i.e. Parkin, PINK 1 and DJ-1 [67, 68].
4. GENES INVOLVED IN THE AUTOSOMAL DOMINANT FORMS OF PD:
4.1. SNCA (PARK1-4)
The product of SNCA is α-synuclein protein. Its aggregation leads to the formation of Lewy bodies [69, 70]. It is localized in presynaptic terminals of the dopaminergic neurons [71]. It is reported that patients suffering from PD with SNCA mutations are associated with early-onset of PD (age of onset ≤50 year). The association of REP1 polymorphism in the promoter region of the gene and PD has been suggested by the researchers [72, 73]. The exact function of α-synuclien is still unknown, but it has been suggested that it plays an important role in the transport of synaptic vesicles thus maintaining the synaptic plasticity [74]. The abnormal form of the protein is more susceptible to fibrillogenesis and self-aggregation [58, 75, 76]. Only three different missense mutations in the form of duplications and triplications have been reported to date [77]. The first missense is p.A53T, which is most frequent and reported in various studies from Italy, Greek, Korea and Sweden [76, 78-82]. p.A53T substitution was discovered in 1997. The p.A30P and p.E46K substitutions were discovered in 1998 and 2004, respectively [53, 76].
The penetrance of the missense mutations is about 85% for p.A53T [83, 84]. However, the penetrance of SNCA duplication was surprisingly found to be low as 33.3%, in Japanese family [85]. The SNCA consist of six exons ranging in size from 42 to 1110 bp and the α-synuclein is about 140 amino acid long [86]. It has three domains: (i) the amino-terminal region (ii) a central hydrophobic domain and (iii) an acidic, negatively charged C-terminal domain. All three missense mutations are associated with the amino-terminal domain [87]. When associated with the phospholipid membranes, via its amino-terminal repeats (amino acids 71-82), it adopts a structure rich in α-helical character [88]. The mutation in the gene results in the formation of toxic oligomers of stable β-sheets and thus forming protofibrils and amyloid fibrils [89-91]. The accumulation of glucocerebrosides, due to the loss of functional glucocerebrosidase enhances the aggregation of α-synuclein. α-synuclein inhibits the lysososmal activity of glucocerebrosidase in the neurons [92, 93].
4.2. LRRK2 (PARK8)
LRRK2 (leucine-rich repeat kinase-2), is a large multidomain protein, involved in neurite outgrowth, synaptic morphogenesis, protein cargo trafficking, autophagy and protein synthesis [94]. The LRRK2 is also involved in the making of dardarin [95]. LRRK2 also play an important role in regulating the innate immune system. It has a conserved supra domain consisting of a Ras-like GTPase called Roc (Ras of complex proteins) and a C-terminal of Roc (COR) domain [96]. LRRK2 is involved in dual enzymatic functions (GTPase and kinase activity). So far 80 mutations have been reported for LRRK2, and only six mutations have been found to be associated with PD i.e. N1437H, R1441 G/H/C, Y1699C, I2012T, G2019S, and I2020T [97-99].
Mutation in the LRRK2 (PARK8) gene (leucine-rich repeated kinase-2) is the most common cause of late-onset autosomal-dominant PD (ADPD) as well as the sporadic form of PD, having a mutation frequency of 2 to 40% among different populations [100-102]. LRRK2 consists of 51 exons and encodes the 2527 amino acid cytoplasmic protein. It consists of a leucine-rich repeat towards the amino terminus and a kinase domain towards the carboxyl terminus. More than 50 different missense and nonsense mutations have been reported for LRRK2 to date among which 16 of them (including the six recurrent mutations- p.R114C, p.R1441G, p.R1441H, p.Y1699C, p.G2019S, and p.I2020T) seems to be pathogenic [103]. The mutation c.6055G >A (p.G2019S) has been reported in 40% of Arabs [101], 20% of Ashkenazi Jews [102], and 1%-7% of Europeans [104, 105]. Few researchers have described p.G2019S mutation in homozygous conditions [106]. Due to the founder effect, p.R1441G is very frequent among Basques [107, 108] and p.I2020T among Japanese patients [109]. The p.G2019S showed reduced penetrance, whereas p.R1441 mutation has been reported to be highly penetrant [110]. The exact mechanism leading to PD by LRRK2 mutations is not properly understood. However, due to its large size, any change in its domain would lead to the loss of its interaction with other proteins, thus inhibiting its function. In addition, various mutations have also been reported to affect its kinase activity [111].
4.3. VPS35, EIF4G1, DNAJC13 and CHCHD2
VPS35, EIF4G1, DNAJC13, and CHCHD2 are the most recent genes to be associated with autosomal dominant form of PD. VPS35 (vacuolar protein sorting 35), a multi subunit complex, is found to be associated with endosomes and is involved in intracellular membrane-bound compartments transportation between the plasma membrane to golgi bodies [112, 113]. EIF4G1 (eukaryotic translation initiation factor 4-gamma) mutations are also linked with PD. It may interact with FMR1, involved in the regulation of the actin cytoskeleton pathway, have shown the strongest association with PD [103, 114]. DNAJC13 [mammalian homolog to receptor-mediated endocytosis 8 (REM-8)] is a chaperone protein found in endosomes and regulates trans-membrane protein transfer. A mutation in DNAJC13 showed aberrant endosomal confinement of α-synuclein, which might stimulate the process of neurodegeneration leading to PD [115, 116]. CHCHD2 (Coiled-coil-helix-coiled-coil-helix domain containing protein 2), is a mitochondrial protein, promotes mitochondrial oxygen consumption and its mutation leads to apoptosis and production of reactive oxygen species (ROS) (mitochondrial dysfunctioning) [117-119].
5. GENES INVOLVED IN AUTOSOMAL RECESSIVE FORMS OF PD
Three genes (parkin, PINK1 and DJ1) have been identified to be associated with autosomal recessive form of inherited parkinsonism. parkin encodes for E3 ubiquitin ligase, which controls the proteasome-dependent degradation or autophagy in neurons [120]. PINK1, is a mitochondrial protein kinase plays an important role in mitophagy [121, 122]. The mutations in DJ-1 bring changes in mitochondrial morphology and leads to the high production of reactive oxygen species [123]. The most common cause of autosomal recessive PD is the mutation in parkin. Parkin mutation has been reported in about 49% of familial cases and about 15% of sporadic cases [124, 125]. Mutations in DJ1 (1-8%) and PINK1 (1-2%) are less common. The function of DJ-1 is not well understood, but it protects mitochondria against the oxidative stress [126-128]. The proteins encoded by parkin, PINK1 and DJ-1 are directly or indirectly involved in the recovery of damaged mitochondria [129-131]. The other genes associated with PD are ATP13A2 [132, 133], FBXO7 [134, 135], PLA2G6 [136, 137], POLG1 [138, 139] and SYNJ1 [140, 141]. Genetic mapping methods such as linkage analysis and genome-wide association studies have been reported to be helpful in identifying new genes responsible for PD [142].
5.1. Parkin (PARK2)
Parkin gene was discovered in 1998. It contains 12 exons.The clinical phenotypes of Parkin, PINK1 and DJ-1 linked PD are identical [143, 144]. A number of parkin mutations have been identified in all 12 exons [106, 124]. The mutations associated with parkin lead to the early onset of PD that accounts for about 77% of the familial cases with an age of onset <30 years [124]. The protein has a conserved Ubiquitin- Like domain (UBL) with Really Interesting New Gene (RING) finger motifs [145, 146]. There are two main targets for parkin (i) CDCrel - 1, belongs to the family of GTPases called septins, and is expressed in synaptic vesicles [145, 147], (ii) Pael-R (Parkin associated endothelin receptor). Parkin specifically ubiquitinates this receptor in the presence of ubiquitin-conjugating enzymes and promotes the degradation of insoluble Pael-R, resulting in the suppression of cell death. In the absence of functional parkin, Pael-R accumulates in the brains and leads to neuronal death [148, 149]. It has been reported that protein synphilin-1, ubiquitinated by parkin leads to the formation of protein aggregates [150].
5.2. PINK1 (PARK6)
PINK1 (PTEN-induced kinase 1), is a mitochondrial serine or threonine-protein kinase. PINK1 is a 581 amino acid protein kinase [151].The mitochondrial membrane protein PINK1 mutations are also found in the forms of familial PD. An amino-terminal of the protein has 34 amino acids (mitochondrial targeting motif), and the carboxy-terminal serve as an autoregulatory domain. Some recent studies have suggested that PINK1 and parkin, are associated with a common pathway, for eliminating the deformed mitochondria [152]. PINK1 attaches itself to the mitochondrial membrane and recruits parkin from the cytosol. Once recruited parkin becomes enzymatically active and initiates the process of eliminating the mitochondria (mitophagy) [153].
Mutations in the phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1) leads to the onset of early autosomal recessive PD. The frequency of PINK1 mutations is about 1-9%, varying among different ethnic groups [129, 151, 154-156]. PINK1 mutations have been reported as either missense or nonsense mutations in the Sudanese family (exons 4-8) [157], exons 6-8 deletion in a sibling family [156], and exon-7 deletion [158], and one with a splice site mutations [159]. To date, about 60 different missense and nonsense mutations have been reported in more than 170 patients, affecting all eight PINK1 exons with a nearly equal frequencies (5-10 different mutations in each exon) [160].
5.3. DJ-1 (PARK7)
The mutation of DJ-1 is reported in about 1-2% of PD cases [161]. DJ-1 has different mechanisms to protect dopaminergic neurons against neurodegeneration in PD [162]. DJ-1-linked PD is rare and so far, only ten different point mutations and exonic deletions have been found to be associated with the disease [163]. The level of oxidative stress regulates its activity [164]. The gene consists of seven exons and codes for a protein of 189 amino acids [165]. In physiological condition, DJ-1 protein forms a dimeric structure [166].
6. OTHER GENES RESPONSIBLE FOR PD
Mutations in ATP13A2 are associated with an autosomal recessive form of PD i.e. Kufor-Rakeb syndrome (KRS). It has a juvenile onset with rapid progression, followed by dementia, supranuclear gaze palsy and pyramidal signs [167, 168]. The gene consists of 29 exons, which codes for a protein of 1180 amino acids. The ATP13A2 protein is normally located in the membrane of lysosome, with 10 trans-membrane domains having an ATPase domain [169]. The first mutation in ATP13A2 (PARK9) was reported in a Chilean family [170, 171]. The lysosomal enzyme β-glucocerebrosidase is coded by GBA gene and play a role in glycolipid metabolism. Mutations in the GBA gene results in the production of non-functional β-glucocerebrosidase which leads to Gaucher disease [172]. GBA mutations are reported to enhance the progression of PD and about 8-14% of the autopsy revealed its link with PD [173-175].
The complex interactions of environmental and genetic factors may lead to PD. Such cases have been classified as sporadic [176, 177]. Damage in the substantia nigra has been reported in most of the sporadic cases [178, 179]. Some gene mutations can lead to the accumulation of unwanted protein that may alter the function of mitochondria leading to the increase in oxidative stress [180, 181]. Table 2 gives a description of genes associated with PD.
| S.No. | Gene with Symbol |
Locus/ Loci |
Chromosomal Location |
Year of Discovery |
Recessive or Dominant | Clinical Features in PD | Reported Variants associated with PD | Target in Cell |
Possible Function(s) |
Reference(s) |
| 1. | α-synuclein (SNCA) | PARK1 and PARK4 | 4q21 | 1997 | Dominant | Hyposomnia, and sleep disorder | rs2583988, rs356219, and rs2736990 | Presynaptic terminals | Regulating synapse, vesicle trafficking | [181, 182] |
| 2. | Parkin (PRKN) | PARK2 | 6q25.2-q27 | 1998 | Recessive | Hyper-reflexia | S167N, E310D, D394N | Mitochondria | E3 Ubiquitin Protein Ligase | [183, 184] |
| 3. | UCHL1 | PARK5 | 4p13 | 1998 | Dominant | Multiple tumors | I93M and S18Y | Nerve cells | Synthesize an enzyme called ubiquitin carboxyl-terminal esterase L1 | [72, 185] |
| 4. | DJ1 | PARK7 | 1p36 | 2003 | Recessive | Dyskinesia, rigidity, and tremors, | C53A and DJ-1C106 | Nerve cells | Transcriptional regulation, antioxidative stress reaction, and chaperone, protease, and mitochondrial regulation | [164, 165] |
| 5. | PINK1 | PARK6 | 1p36-1p35 | 2004 | Recessive | Dystonia | P37L, R42P, R256C, G328E, and R334C | Mitochondrial membrane | Mitochondrial kinase | [186, 187] |
| 6. | POLG | POLG1 | 15q26.1 | 2004 | Dominant | Bradykinesiaand rigidity, resting tremor and they respond well to levodopa treatment | p.A899T | Cortex | CAG-repeat, encoding a polyglutamine (poly-Q) trac | [188-190] |
| 7. | LRRK2 | PARK8 | 12q12 | 2004 | Dominant | Less non-motor function, olfactory, and cognitive impairment | A419V, G2019S, R1441C/G/H, G2385R, and R1628P | Glial cells and Neurons | Protein kinase | [191-192] |
| 8. | HtrA2 | PARK13 | 2p12 | 2005 | Dominant | Bradykinesia, tremor | Omi/HtrA2, Pro143Ala | Glia cells and Neurons | Mitochondrial serine kinase | [193-195] |
| 9. | ATP13A2 | PARK9 | 1p36 | 2006 | Recessive | Kufor-Rakeb syndrome, pallido-pyramidal syndrome, dystonia | F182L and G504R | Lysosomes | Improves the lysosome membrane integrity and protects against iron-induced cell damage | [132, 196, 197] |
| 10. | FBOX7 | PARK15 | 22q12-q13 | 2008 | Recessive | Rigidity, bradykinesia and postural instability | Arg498Stop | Cerebral cortex, Globus pallidum and the Substantia nigra | Ubiquitin ligase | [134, 198] |
| 11. | GIGYF2 | PARK11 | 2q37.1 | 2008 | Dominant | Less motor symptom | N56SN457Trs12328151, rs2289912, rs2305138, rs3816334, A572AH1171R | Endosomes | Progression in motor symptoms | [199, 200] |
| 12. | GBA | GBA1 | 1q21 | 2009 | Dominant | Hallucinations and cognitive decline | E326K, T369M, N370S, D409H, and L444P | Endoplasmic reticulum | Regulation of the lysosomal system | [201, 202] |
| 13. | PLA2G6 or iPLA2β | PARK14 | 22q12-q13 | 2009 | Recessive | Axonal dystrophy, dystonia, dementia, visual disturbances | p.R301C and p.D331N | Peripheral nerves | Phospholipase A2 | [203, 204] |
| 14. | Vps35 | PARK17 | 16q11.2 | 2011 | Dominant | Tremors, bradykinesia, rigidity of limbs and gait defects | p. Asp620Asn | Lysosomal vesicles | Retromer component | [205-207] |
| 15. | EIF4G1 | PAERK18 | 3q27.1 | 2011 | Dominant | Motor dysfunction | A502V, G686C, S1164R, and R1197W | Presynaptic nerves | Scaffold subunit of the translation initiation complex eIF4F, which binds the ribosomal 40S | [114, 208] |
| 16. | DNAJC6 | PARK19 | 1p31.3 | 2012 | Recessive | Mental retardation, pyramidal signs and epilepsy | p.Q734X | Cerebellum, corpus callosum, cortex, brainstem, pons, putamen, spinal cord, and substantia nigra. | Cochaperone | [209, 210] |
| 17. | SYNJ1 | PARK20 | 21q22.11 | 2013 | Recessive | Dystonia, oculomotor apraxia, and seizures | Nil | Presynaptic terminal | Polyphosphoinisitide phosphatase | [211, 212] |
| 18. | DNAJC13 | PARK21 | 3q22.1 | 2014 | Recessive | Motor dysfunction | p. N855S | Neurons | Endosomal transport | [213, 214] |
| 19. | CHCHD2 | CHCHD2 | 7p11.2 | 2015 | Recessive | Bradykinesia and rigidity, hypomimic face and mydriatic pupils | G66V and P80L | Cortical neurons | Mitochondrial protein | [215, 216] |
| 20. | LRP10 (low-density lipoprotein receptor-related protein 10) | LRP10 | 14p13–q12 | 2018 | Dominant | Motor fluctuations and dyskinesia, no cognitive impairment | OR11H12, POTEG, and LRP10 | Cerebral cortex | Regulates amyloid precursor protein (APP) trafficking and processing. | [217-219] |
7. POST TRANSLATION MODIFICATIONS OF SNCA, DJ1, PARKIN AND LRRK2
The pathogenesis of PD mainly involves dysfunction of mitochondria, protein misfolding, aggregation and oxidative stress [220]. A number of post-translational modifications (PTMs) regulates the activity of protein; for example, nitration, arginylation and N-terminal acetylation which affects the structure and behaviour of α-synuclein to interact with other biomolecules [221]. The modification of α-synuclein, its aggregation, toxicity and targets for future therapeutics has been extensively reviewed by Barrett and Greenamyre [222]. Although PTMs are required for the normal functions of α-synuclein, abnormal modifications may promote pathogenesis of the PD [223]. The phosphorylation of S129 residue has been reported to be associated with the formation of Lewy bodies [224]. Nitration of tyrosine residues in α-synuclein at Y39, Y125, Y133 and Y136 has been reported to be associated with oxidative stress [225]. Environmental factors are also responsible for the PTMs. The exposure of 1-Methyl-4-phenyl-1,2,3,6-Tetrahydropyridine (MPTP) results in the phosphorylation of S-129 in α-synuclein [226]. It has been observed that Mn induced the aggregation of α-synuclein and the similar effect was observed in cultured cells exposed to fungicide Maneb (having Mn) showed the same results [227].
Parkin, Pink1 and DJ-1 are involved in the formation of the heteromeric cytosolic complex (PPD complex) which is involved in Ubiquitin mediated proteasomal degradation [228]. It has been reported that Parkin can mediates its own ubiquitylation and give rise to high molecular mass k49-linked polyubiquitlylated moiety [229]. Phosphorylation at two different sites (T175, T217) was reported, but has not been confirmed by other researchers [230]. There are evidences that the activity of Parkin, choice of E2, choice of substrate, localization and solubility can be regulated by post-translational modification [231]. It has been reported that reactive cysteine (cys106) is essential for the proper functioning of DJ1 in humans [232]. Due to the increase in oxidative stress either by aggregation of α-synuclein or environmental agents promotes the oxidation of cysteine to sulfenic, sulfinic and sulfonic acid [232]. The oxidation results in the loss of the activity of DJ-1 and thus enhances the progression of PD. The phosphorylation of LRRK2 can occur in two ways constitutive and autophosphorylation. These phosphorylations are modulated in PD [233]. The mutations in the ROCO domain (Ras of complex proteins) results in the decrease of phosphorylation at R1441C, R1441G, and Y1699C, which leads to the aggregation of LRRK2 in the substantia nigra [234]. Ubiquitination also regulates the activity of LRRK2. The mutations R1441C, R1441G and Y1699C and 12020T may result in the decrease of constitutive phosphorylation or hyper-ubiquitination or instability, leading to the formation of protein aggregates [235].
8. GENOME WIDE ASSOCIATION STUDIES (GWAS)
Genome wide association studies are helpful in understanding the genetic basis of PD [236]. About 24 loci have been reported to be associated significantly with the risk of disease in the individuals of European ancestry [237]. In another study, 17 novel PD loci, using a neurocentric candidate gene nomination pipeline, found that these loci play a role in lysosomal biology and autophagy [238]. The identification of such candidate genes will definitely help in understanding the role of causal genes and will also open new horizons for the therapeutic approaches.
9. TREATMENTS FOR PARKINSON’S DISEASE
The past half century of research has brought many alluring advancements in the treatment of PD. It is a fact that PD is a progressive disease with no permanent cure, but the symptoms can be suppressed or delayed by the treatment of levodopa [239-241]. To overcome the side effects of Levodopa, there are several treatment strategies for the management of PD in order to provide more improvement in combating the PD symptoms [242]. Various treatment strategies for the PD are as follows:
9.1. Levodopa
Levodopa is also known as L-Dopa (l-3,4-dihydroxy phenylalanine). For treating early symptoms of PD the levodopa is given to the PD patients [243]. It is a precursor of dopamine which is activated through decarboxylation by the cytosolic enzyme Aromatic Amino Acid Decarboxylase (AADC). It is formed by the hydroxylation of the amino acid L-tyrosine. This reaction is catalyzed by tyrosine hydroxylase and is a rate-limiting step in the biosynthesis of the neurotransmitter dopamine. Dopamine is formed by the decarboxylation of levodopa in the presence of enzyme L-amino acid decarboxylase or Dopa Decarboxylase (DDC). The absorption of levodopa takes place first in the small intestine and enters as large neutral amino acids (LNAAs). Levodopa accesses in brain by the help of LNAAs system, where it is converted to dopamine. Once the dopamine is formed it acts on dopamine receptors (D1 and D2 families) to improve the motor functions in PD. The levodopa is absorbed by the specific LNAAs transporter i.e. protien rich meals. The therapeutic efficacy of levodopa is reduced due to the over-saturation of the transporter with amino acid derived from dietary protein. Thus, it is recommended that the levodopa should be taken without the meal to ensure the full absorption [244, 245]. In commercial preparations, levodopa is combined with carbidopa or benserazide, which act as inhibitors of decarboxylase, thus reducing the risk of peripheral side effects [246, 247].
The long term use of levodopa is associated with side effects such as nausea, vomiting, constipation and hallucinations [248, 249]. L-dopa may also induce cell death in neuronal cells by increasing the activity of caspase-3 [250, 251]. L-dopa treatment may lead to alteration in D1 dopamine receptors and might form the basis for the occurrence of dyskinesia and motor fluctuations [252, 253].
9.2. Catechol-O-methyl Transferase (COMT) inhibitors
COMT is an enzyme that metabolizes or degrades neurotransmitters such as dopamine, hence the COMT inhibitors may be useful to enhance the half-life of dopamine. Tolcapone and entacapone are commonly used in COMT inhibitors. Although, both the drugs are associated with side effects such as diarrhea, hepatotoxicity, increased dyskinesia, and urine discoloration [254, 255]. Opicapone, a third-generation COMT inhibitor, is also used to treat PD at a dosage of 50 mg daily [256]. It is prescribed in Germany and the United Kingdom, but further detailed studies are required for the validation of the efficacy and long-term risks associated with it. Most of the COMT inhibitors prevents the peripheral catabolism of L-dopa and increase the half-life of L-dopa [257, 258].
9.3. Amantadine
Although the exact mechanism of amantadine is not known, it is thought that the symptomatic relief is due to its anticholinergic property [259]. It blocks the glutamatergic N-methyl-D-aspartate (NMDA) receptors in the basal ganglia and thus helps in reducing the dyskinesias associated with L-dopa therapy [260, 261].
9.4. Anti-cholinergic Drugs
Anticholinergics (anti-muscarinics) are used to reduce tremors in PD patients. Due to neuropsychiatric side effects (memory loss, confusion) and autonomic side effects (constipation, urinary retention, dry mouth), these drugs are not normally recommended to the patients above 65 years [262]. Trihexyphenidyl, benztropine, orphenadrine, procyclidine, and biperiden are commonly use anticholinergic drugs [263-265].
9.5. Monoamine Oxidase Inhibitors (MAOIs)
They inhibit the activity of one or both monoamine oxidase enzymes i.e. Monoamine Oxidase A (MAO-A) and Monoamine Oxidase B (MAO-B). They are used as a powerful anti-depressants to treat panic disorders and social phobias [266, 267]. MAO-A shows greater affinity for hydroxylated amine, i.e. noradrenaline (NA) and serotonin (5-HT), while MAO-B shows greater affinity for non-hydroxylated amines, i.e. benzylamine and β-Phenylethylamine (PEA) [268, 269]. MAO-A degrades dopamine, norepinephrine and serotonin by oxidative deamination. It is mostly expressed in neuronal cardiac cells and is localized in the outer mitochondrial membrane [270, 271]. MAO-B enzymes are mostly expressed in the brain and regulate the storage as well as free intra-neuronal concentration of dopamine. MAO-B is involved in the breakdown of dopamine [272, 273]. It has been well established that the inhibition of MAO-B increases the level of synaptic dopamine and provides symptomatic relief [274]. Two commonly used MAO-B inhibitors are selegiline and rasagiline. Selegiline is used as adjunctive therapy to L-dopa for patients exhibiting motor fluctuations [275, 276]. Rasagiline (N-propargyl-1(R)-aminoindan) is a selective
| Ergoline Derivatives | Non-Ergoline Derivatives | ||||||||
| Name of Derivatives | Bromocriptine | Cabergoline | Pergolide | Lisuride | Apomorphine | Piribedil | Pramipexole | Ropinirole | Rotigotine |
| Trade Name (s) | Parlodel and Cycloset | Cabaser and Dostinex | Permax and Prascend | Dopergin, Proclacam and Revanil | Apokyn, Ixense, Spontane and Uprima | Trivastal, Pronoran, Trastal and Trivastan | Mirapex and Mirapex ER | Requip and Requip XL | Neupro |
| Half-life (t½) | 12-14 hours | 63-69 hours | 27 hours | 2 hours | 30-60 minutes | 20 hours | 8-12 hours | 6 hours | 3 hour |
| Peak Plasma Time | 58 minutes | 2-3 hours | 2-3 hours | 40 min | 10-60 min | 1 hour | IR-2 hour, ER-6 hour |
IR -1-2 hour, ER 6-10 hour | 15-18 hour |
| Highest binding affinity | D2 receptors | D2 receptors | D2 receptors | D2 receptor | D2 receptor | D2 and D3 receptors | D3 receptor | D2 and D3 receptors | D3 receptor |
| Chemical formula | C32H40BrN5O5 | C26H37N5O2 | C19H26N2S | C20H26N4O | C17H17NO2 | C16H18N4O2 | C10H17N3S | C16H24N2O | C19H25NOS |
| Protein Binding (%) | 90-96% | 40-42% | 90% | 15% | 50% | - | 15% | 40% bound to plasma proteins | 92% |
| Metabolism | Gastrointestinal tract and liver | Hepatic | Hepatic | - | Hepatic | - | Hepatic | Hepatic | Hepatic |
| Route of Administration | oral, vaginal, intravenous | Oral | Oral | Oral | Subcutaneous | - | Oral | Oral | Transdermal |
| Route of Elimination | Liver and Kidney | Urine, Fecal | Kidney | - | - | - | Urine | Urine | Urine, Fecal |
inhibitor of MAO-B and therefore does not interfere with MAO-A. It has been approved to be used as an adjunct therapeutic agent for the PD patients on L-dopa and experiencing motor fluctuations [277-279]. It has a prolonged effect on the ability to reduce the motor fluctuations associated with L-dopa therapy [280]. Safinamide, an MAO-B inhibitor, has been introduced in Europe as an adjunct therapy to levodopa. It is a selective and reversible MAO-B inhibitor [281-284].
9.6. Dopamine Agonists (DAs)
DAs are synthetic drugs which directly act on striatal postsynaptic dopamine receptors. They are reported to have a better pharmacokinetic profile than levodopa and are divided into two broad classes: Ergot derived and nonergot derived. The ergot-derived includes: Bromocriptine, cabergoline, lisuride, dihydro-ergocryptine, and pergolide [285, 286]. Non ergot-derived includes: Pramipexole, ropinirole, piribedil and rotigotine [287]. Apomorphine is used in advanced stages of PD. It acts on both D1 and D2 dopamine receptors. It displays the pharmacological profile of all clinically available DAs, but due to the short half-life and poor oral bioavailability, it is not used for the treatment of early stages of PD [288]. DAs are also associated with side effects such as nausea, day time somnolence, confusion, hallucinations, leg edema, orthostatic hypotension, and erythromelalgia (particularly ergot derivatives) [285]. The pharmacological properties of ergot and non-ergot derived drugs are listed in Table 3.
10. NONPHARMACOLOGICAL THERAPIES
Non-pharmacological therapies includes: Deep Brain Stimulation (DBS), Gene therapy and Cell transplantation.
10.1. Deep Brain Stimulation (DBS)
In DBS, the two hyperactive regions during the progression of PD i.e. Subthalamic Nucleus (STN) and Globus Pallidus internus (GPi) are used as targets. DBS blocks unusual signals through the cortico-basal ganglia loop [289]. DBS of STN and GPi-DBS showed a significant improvement in dyskinesias and motor fluctuations associated with PD patients [290, 291]. A study on low frequency (60 Hz of DBS) showed great potential to improve overall motor symptoms associated with PD, such as swallowing, gait and freezing [292-294]. Some pre-clinical studies have concluded that DBS showed improvement in the survival of dopaminergic neurons and also the levels of BDNF in the substantia nigra [295, 296]. According to Wong et al. [297], STN-DBS and GPi-DBS are equally potent in reducing tremors associated with PD patients.
10.2. Gene Therapy
Gene therapy involves creating new cells that produces a specific neurotransmitter (i.e. dopamine). The modified cells are transplanted in PD patients [298]. It is well known that enzyme Aromatic Acid Decarboxylase (AADC) plays a major role in converting the levodopa to dopamine. In PD patients, the loss of neurons in the substantia nigra leads to the loss of AADC, which leads to the inability to convert levodopa to dopamine. The gene therapy has been conducted to restore the activity of AADC in the striatum, which levodopa to dopamine [299, 300]. In 2012, the gene therapy experiment was also performed on primates for Tyrosine Hydroxylase (TH) activity in astrocytes. The results showed behavioral improvement in the rats receiving transplantation by gene therapy [301, 302].
10.3. Cell Transplantation
Cell transplantation has been used for decades and the clinical trials have shown positive results improving the motor symptoms [303-305]. In 2015, dopaminergic neurons transplantation was reported to maintain healthy and functional expression of DA transporters with normal mitochondrial morphology [306].
10.4. Physical Therapy
Physical therapy plays an important role in improving walking and postural instability. The physical therapy includes some compensatory strategies such as, motor skill learning and education to optimize physical activity. According to Morris et al. [307] management of gait disorders in people with PD should follow a critically designed training. In this training PD patients should be taught how to move more easily and maintain postural stability by using cognitive strategies. It targets the primary motor control deficits in the basal ganglia, brain stem, and motor cortex. There are two forms of strategies: (1) compensatory strategies to pass the defective basal ganglia and (2) learning strategies to improve performance through practice. Worldwide, Physical therapists provides the following measures to reduce the PD symptoms [308]:
- Lecture on self-management.
- Routine exercise associated with improvements in mobility, quality of life and disease severity.
- Ways to maintain safety while performing exercises.
- Help in normal physical activity which includes walking.
- Practicing to maintain balance or stability.
- Getting around (daily routine work).
CONCLUSION
Since there is no permanent cure for the disease to date, a continuous efforts in the direction of treatment strategies is the need of the hour. It is difficult to differentiate between the sporadic and familial forms of PD on the basis of symptoms. Recently, the GWAS studies have paved the pathway for studying the collective role of the loci in the progression of PD. Hence, GWAS should be conducted for the better understanding of pathology as well as treatment of PD.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

